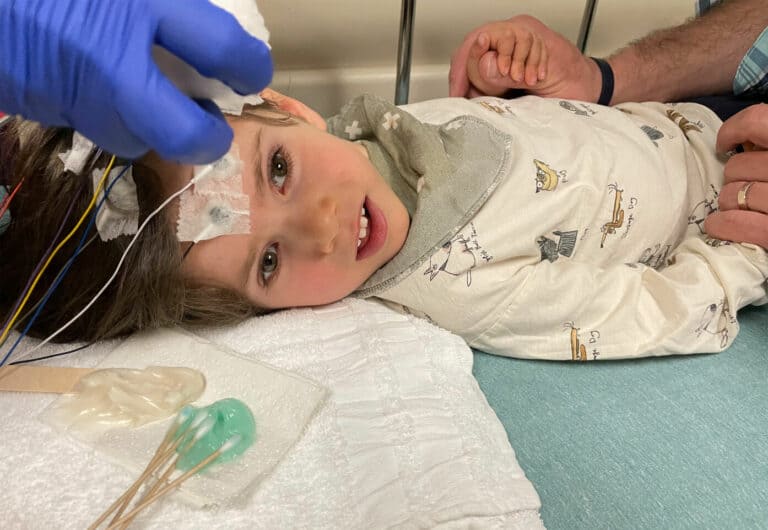
عندما يصلي غاري لاندسمان، يتخيل أن كلاً من ولدَيْه بيني وجوش يركضان نحوه وهما يرتديان قبعات اليارمولك الصغيرة والجدائل والأشرطة القطنية المتدلية من حزام الخصر التي تسمى تسيت سيت، ويفتح ذراعيه استعداداً لاحتضانهما.
ولكن الحقيقة هي أن بيني وجوش يعانيان من داء كانافان، وهو اضطراب دماغي وراثي مميت. فهما مثبّتان في كرسيهما المتحركين، ولا يمكنهما الكلام أو التحكم في أطرافهما.
وفي يوم الخميس الموافق 8 أبريل، قام لاندسمان وعائلته بإدخال الولد الأكبر بيني إلى مستشفى في مدينة دايتون بولاية أوهايو، حيث قام جراحو الأعصاب على مدار عدة ساعات بحفر ثقوب في جمجمته وحقن تريليونات من الجسيمات الفيروسية التي تحمل النسخة السليمة من جين يفتقده جسمه.
تمثل هذه العملية ذروة سعي دام أربع سنوات من قبل عائلة لاندسمان، التي تعيش في بروكلين بولاية نيويورك، للحصول على علاج جيني، تعتقد العائلة أنه الأمل الوحيد لإنقاذ طفليها.
كشفت إم آي تي تكنولوجي ريفيو عن قصة عائلة لاندسمان لأول مرة في قصة غلاف العدد الخاص بالطب الدقيق في عام 2018. تسمح التطورات في تقنيات العلاج الجيني بمعالجة الأمراض الوراثية مثل الهيموفيليا. ولكن نظراً لأن داء كانافان هو مرض نادر للغاية، فإن القليل من الشركات تعمل على علاجه. لذا قامت الأسرة بتمويل العلاج الجيني الجريء بنفسها، وذلك باستخدام الأموال التي جمعتها عبر الإنترنت.
إن التقدم المذهل في تقنيات تسلسل الجينوم واستبدال الجينات وتعديلها يعني نظرياً إمكانية علاج آلاف الأمراض الوراثية النادرة. ولكن يقول والدا الطفلين إن كون الشركات لا تتصدر هذا الاتجاه، فقد اضطرا إلى طلب ملايين الدولارات لتمويل التجارب المطلوبة. كما أن هناك المعضلة الأخلاقية؛ ففي بعض الحالات، يقوم الوالدان بجعل أطفالهم هم أول من يتلقى العلاج.
فعلى سبيل المثال، تعطي التجربة في مدينة دايتون الأولوية للأطفال الذين تمكنت عائلاتهم من جمع الأموال للقيام بالتجربة، التي تقترب تكاليفها حتى الآن من 6 ملايين دولار. وتقول أليسون بيتمان هاوس، الأخصائية في الأخلاقيات الحيوية في جامعة نيويورك والتي تدرس القضايا الأخلاقية في تجارب العلاج الجيني للأطفال: "يثير الأمر السؤال الدائم المتعلق بالعدالة حول من يمكنه الوصول إلى التجارب ومن لا يمكنه ذلك".
جمعت عائلة لاندسمان أكثر من مليوني دولار، كما اعتمدت عائلات من روسيا وبولندا وسلوفاكيا وإيطاليا على التبرعات النقدية لضمان مشاركتها في التجربة. ونشرت عائلة روسية نسخة من فاتورة مخصصة لـ "العلاج الجيني" بمبلغ مليون و140 ألف دولار، التي تضمنت 800 ألف دولار مقابل تكاليف تصنيع العلاج الجيني المستخدم في التجربة.
ووفقاً للدعوة العاجلة التي أطلقتها الأسرة الروسية لجمع التبرعات، فإن فشلهم في دفع هذا المبلغ سيؤدي إلى خسارة طفلتهم الصغيرة أولغا "لفرصتها الوحيدة للشفاء، وذلك من خلال علاج مكلف في الولايات المتحدة". وانتهى بهم الأمر عبر المساهمة بما لا يقل عن 700,000 دولار.
وفي حين أن هذه التجارب "التي يتم المشاركة بها مقابل الدفع" قانونية، إلا أنها تثير مسائل إشكالية، بما فيها المسألة المتعلقة بما إذا كان الآباء والأمهات والمتبرعون بالمال يدركون أن معظم العلاجات التجريبية تواجه الفشل. وتقول بيتمان هاوس: "ليست بالضرورة غير أخلاقية، ولكن يجب التدقيق على سبب مطالبة المريض بالدفع. إذا كانت التجربة قابلة للتطبيق، فلماذا لا تهتم بها المعاهد الوطنية الأميركية للصحة أو إحدى شركات التكنولوجيا الحيوية؟ لماذا لا يوجد هناك مصدر آخر للتمويل؟".
هل سينجح الأمر؟
يحدث داء كانافان عندما يرث الطفل نسختين شاذتين من جين يسمى ASPA. دون الإنزيم الذي ينتجه الجين ASPA، لا يمكن للدماغ تشكيل الحزم العصبية التي تنقل الإشارات فيه بشكل صحيح. والنتيجة هي أن بيني وجوش لا يمكنهما التحدث أو التحكم في أطرافهما، فضلاً عن محدودية إدراكهما.
تقول باولا ليون، الباحثة في جامعة روان في نيوجيرسي، والتي ابتكرت العلاج الجيني وقادت الجهود لبدء التجربة السريرية: "إنهم مثل الأطفال الرضع ولكن بأجسام مصابة بالتصلب الجانبي الضموري".

الصورة منشورة بإذن من جيني لاندسمان

الصورة منشورة بإذن من جيني لاندسمان
تسعى التجربة في مستشفى دايتون إلى استخدام الفيروسات لإيصال نسخ وظيفية من جين ASPA إلى أنسجة دماغ الأطفال. وهذا ما حدث مؤخراً في مستشفى دايتون للأطفال. فبعد أن تم الترحيب ببيني من قبل كلب أليف مهمته تشجيع المرضى، قام جراحو الدماغ بحفر ثقب في جمجمته ثم استخدموا إبرة لإدخال 40 تريليون جزيء فيروسي.
تعتمد حجة ليون العلمية على أن إضافة نسخ سليمة من جين ASPA إلى خلايا دماغية خاصة تسمى الخلايا الدبقية قليلة التغصن قد توقف تقدم المرض، وربما تسمح بالتعافي إلى حد ما. وتقول إن العلاج كان فعالاً لدى الفئران، ولكن "هل سينجح الأمر عند المرضى؟ الطريقة الوحيدة هي تجربته".
الآباء والأمهات يصبحون كالعلماء
يوجد الآن ستة أمثلة على علاجات جينية تم تمويلها من قبل عائلات تهدف إلى علاج أطفالها، ويتم التخطيط لإجراء المزيد من هذه التجارب. حتى أن العلماء بدأوا يطورون دواءً فائق التخصيص مصمماً على وجه الخصوص للأطفال الذين يعانون من مشاكل جينية فريدة.
إن هذه الجهود اليائسة تجعل الآباء يتغلبون على عقبات شبه مستحيلة؛ إذ يجب أن يصبحوا خبراء في تطوير الأدوية وأن يجمعوا الملايين وأن يتواصلوا مع العلماء بلا كلل لإقناعهم. ولا يمكن أن يقوم بذلك سوى قلة من الناس.
يقول ساناث كومار راميش، مطور برامج يعاني ابنه من مرض نادر آخر: "هناك الكثير من الأشخاص الذين يعرفون كيفية إجراء العلاج الجيني، ولكن المعرفة مجزأة، ويمكن أن يحدث الكثير من الأخطاء". أسس راميش مؤسسة أوبن تريتمنتس (Open Treatments)، التي تعمل على إنشاء برامج يمكن أن تستخدمها العائلات لتنظيم أبحاث العلاج الجيني، بما في ذلك بعض الخطوات مثل توظيف علماء لإنشاء نماذج حيوانية لمرض ما.
ويقول: "أعتقد أن التمييز بين العلماء والآباء سيكون غير واضح في المستقبل".
بالنسبة للآباء والأمهات الذين تم قبول أطفالهم في تجربة مستشفى دايتون، قد يكون العلاج الجيني هو فرصتهم الأخيرة. من بين أولئك الأمهات كانت ميغان روكويل، وهي فنية متخصصة في الأظفار في مدينة سيدار رابيدز بولاية أيوا، والتي تم في عام 2018 تشخيص إصابة ابنتها توبين غريس البالغة من العمر الآن ثلاث سنوات ونصف بداء كانافان.
وتقول روكويل: "قالوا لنا للأسف، ليس هناك ما يمكننا القيام به. لا يوجد أي إجراء علاجي أو شافٍ، وستكونون محظوظين إذا بلغت عيد ميلادها الخامس. لقد كان الأمر قاسياً أن تعرف أن طفلك الوحيد يعاني من مرض دماغي سيودي بحياته".
وتقول روكويل إنها اكتشفت محاولات ليون لتطوير علاج جيني عن طريق الإنترنت وجمعت في النهاية أكثر من 250 ألف دولار. وتقول: "في ذلك الوقت، كانت توبين أصغر شخص يصاب بداء كانافان في الولايات المتحدة، وأعتقد أن ذلك لعب دوراً كبيراً في قبولها"، مضيفةً أن ليون أخبرت الآباء والأمهات بأن الأموال تجعلهم في مقدمة الطابور ولكنها لا تضمن العلاج.
وتقول بيتمان هاوس، عالمة الأخلاقيات الحيوية، إن هناك خطراً آخر يتمثل فيما إذا كان الآباء والأمهات يستطيعون حقاً الحكم على فوائد الإجراء التجريبي بطريقة "غير عاطفية"، خاصةً إذا وضعوا ثروة كبيرة في هذا المسعى. وتقول: "لا يقتصر الأمر على أن طفلهم يعاني من حالة خطيرة، بل إن تمويل التجربة يأتي من خلال تعبهم وبكائهم. قد يكون من الصعب للغاية على الآباء والأمهات تغيير رأيهم والقول بأنهم لن يفعلوا ذلك".
الأمل مقابل المخاطرة
تشتمل دراسة مستشفى دايتون حالياً على كميات من العلاج الجيني تكفي لعلاج 9 أو 10 أطفال فقط. تم تصنيع العلاج في إسبانيا، ولكن لم يتم ذلك إلا بعد أن تجاوز الباحثون والعائلات ما أطلقوا عليه الإجراءات الروتينية المفرطة والتأخيرات والعقبات، التي وضعت بعضها الهيئات التنظيمية الحكومية التي تحدد العلاجات الجينية التي يمكن تجربتها وما إذا تم التخطيط للتجارب بشكل صحيح.
في عام 2019، اصطحبت عائلة لاندسمان ابنيها إلى إدارة الغذاء والدواء الأميركية لحضور اجتماع تم عقده بعد عشرات المكالمات مع المشرّعين. وتقول والدة الطفلين جيني لاندسمان: "قبل ذلك، كنا مجرد حالة لها رقم وسط كومة كبيرة لديهم من الأوراق. كان لديهم اعتراضات تقنية للغاية. حملنا في الاجتماع بيني وجوش وقلنا بأننا نأمل ألا تؤدي هذه المشكلة التقنية للغاية إلى إيقاف العلاج".
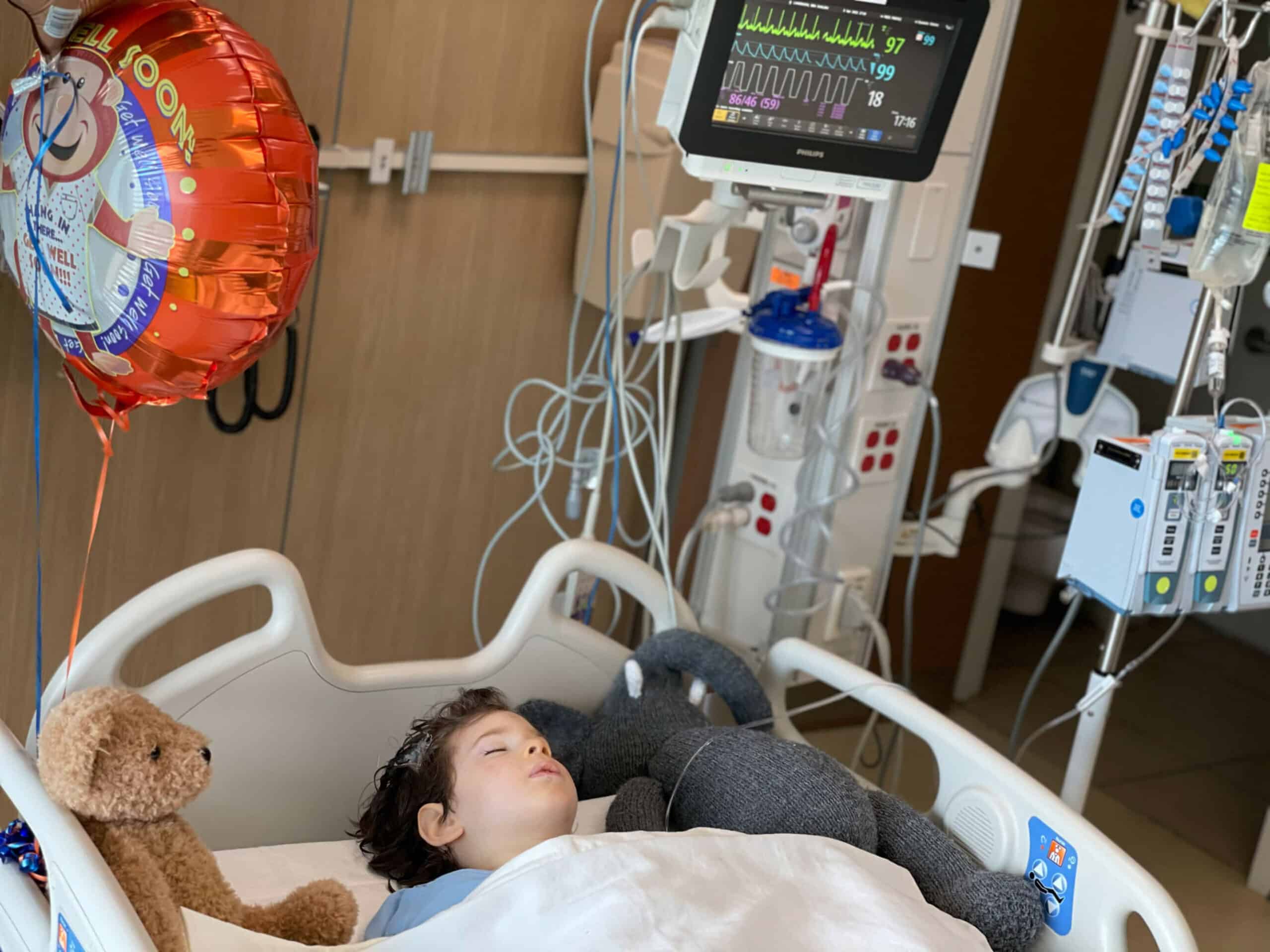
الصورة منشورة بإذن من جيني لاندسمان
حصلت تجربة مستشفى دايتون على الضوء الأخضر في شهر ديسمبر وبدأت في الوقت المناسب تماماً لبيني، الذي سيصل إلى حد الخمس السنوات في شهر يونيو. تقول روكويل، التي ليس لديها حتى الآن موعد لعملية ابنتها: "يمثل بيني العملية التجريبية. بيني هو الطفل الذي نأمل أن تنجح عمليته".
ما هو احتمال نجاح العلاج؟ حققت تقنيات استبدال الجينات نجاحاً ملحوظاً في علاج الأطفال الذين ليس لديهم جهاز مناعي وفي الوقاية من أمراض الدماغ. ومنذ عام 2017، تمت الموافقة أيضاً على تسويق عدد قليل من العلاجات الجينية في الولايات المتحدة، بأسعار مرتفعة تصل إلى 2.1 مليون دولار لكل طفل.
أثارت الأسعار القياسية اهتمام شركات التكنولوجيا الحيوية المتخصصة، التي ترى الآن أرباحاً تجارية حتى من الأمراض النادرة للغاية. تقول إحدى هذه الشركات التي تدعى أسبا ثيرابيوتكس (Aspa Therapeutics)، إن لديها خططاً لبدء تجربة لعلاج جيني مختلف لداء كانافان. ويقدّر الرئيس التنفيذي إريك ديفيد بأن هناك 1,000 طفل يعانون من المرض في الولايات المتحدة وأوروبا. ويقول: "يعدّ ذلك كافياً بالنسبة لنا".
لا يوجد ضمان لنجاح العلاج الجيني مع داء كانافان. فحتى لو أوقف الجين السليم تقدم المرض، فقد تكون أدمغة الأطفال قد تضررت بشكل غير قابل للإصلاح.
تقول روكويل عن ابنتها: "آمل أن تجلس بمفردها، وربما أن تقول ماما وبابا. أنا متفائلة، لكن الأمر تجريبي بحت. نحن نقدّم أطفالنا للعلم ونأمل أن ينجح الأمر وندعو أن يحصل ذلك". لن يعرف الأطباء قبل مرور شهر ما إذا كان الجين الجديد يقوم بوظيفته في دماغ بيني، ولكن من المرجح أن تستغرق معرفة تأثيره على الأعراض وقتاً أطول بكثير.
وفي رسالة إلى المتبرعين، تناول غاري لاندسمان ما أسماه السؤال "الثقيل" لما يتوقع من العملية أن تحققه.
وكتب: "لقد فكرت في هذا السؤال مراراً وتكراراً. هل من المقبول أني أريد المزيد؟ هل من المقبول أني أريد الإمساك بأيديهما وهما يسيران إلى جانبي؟ هل من المقبول أني أريد أن أسمعهما وهما يتحدثان معي؟ ربما أجازف مجازفة خطرة مع نفسي. لكنني أعتقد أن الأمل الذي يوفره الأمر يستحق المخاطرة".